WORK INFORMATION
BIOMS - Center for Modelling and Simulation in the Biosciences
MCM group at EML Research gGmbH
Modeling macromolecular motions in the cell by Brownian dynamics simulations
Macromolecular motions and interactions in the cell are essential steps
in cellular life and occur on a variety of time scales. Processes like
macromolecular diffusion and transport, many types of protein-protein
interactions, and protein domain rearrangement occur on timescales of
milliseconds and longer. These processes cannot be described by
standard molecular dynamics (MD) simulation methods. Brownian dynamics
(BD) simulation is one of the methods, that allows to simulate
macromolecular motions on the millisecond time scale keeping atomic
level accuracy in the representation of the molecules, although not
taking into account internal dynamics of macromolecules. The motions of
solute molecules or particles can be described as Brownian motions,
when solvent molecules are not taken into account explicitly but their
influence on the solute is taken into account via random forces and
friction. As a result, BD simulations model macromolecular motions with
low resolution (~1 A) and larger timesteps (~ 1 picosecond for proteins
under normal viscosity conditions) compared t MD simulations. Larger
time scale (milliseconds) processes can be modeled by BD simulations
with rigid proteins as compared to molecular dynamics simulations of
macromolecules (typically nanoseconds).
The purpose of the project is to do Brownian dynamics simulations of
the interaction of macromolecules using a detailed force field
including hydrophobic interactions. It is planned that the simulation
programs will be developed together with necessary force field
parameters, relevant to the investigated cases of protein interactions.
Currently, the work is being completed on calibrating hydrophobic
interactions on the basis of the simulations of electron transfer
proteins. These proteins form only short living complexes, so that a
full course of their interaction can be simulated at a reasonable
computational cost. Fast and accurate enough method computing
hydrophobic forces is implemented. These forces are proportional to the
changes of solvent accessible areas of proteins in the course of their
interaction. The electron transfer between donor and acceptor sites of
the proteins is modeled in details as a combination of contributions
from many different possible pathways along the atomic bonds and
through solvent.
Electron transfer proteins interact within millisecond time scale and
simulations reproduce this time scale. Simulation results correlate
with available experimental data on electron transfer rates for
different proteins and under different conditions. One of the
interesting results is that, in all simulated cases, the diffusion
contributes to the electron transfer process only partly. A significant
contribution comes from the activation barriers of electron transfer
events at conformations met during diffusion. The situation is very
different from the case, when the proteins are bound tightly for a long
time.
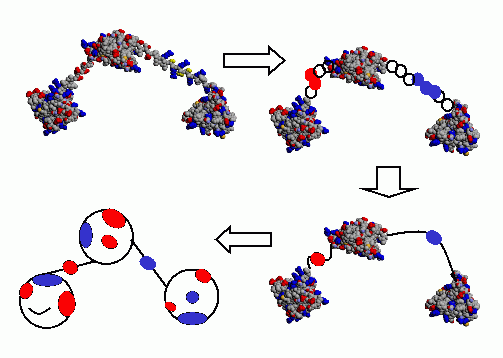
The work is being done on deriving interaction properties of reduced
models of proteins from interaction properties of all-atom models. The
reduced models are needed to do thesimulations extending to hundreds of
milliseconds. It is important to deal with realistic magnitude and
spatial range of interactions, while the proteins theirselves can be
represented as spheres. This is especially important because our BD
simulations are designed to be conforming with multi-scale simulation
methodology, where different simulations methods (molecular dynamics of
all atoms Brownian dynamics of rigid proteins diffusion of
spherical models) can be used one after another depending on the stage
of the process under investigation.
Related Research
SDA (Simulation of Diffusional Association of proteins) version 4.23
ProSAT (Protein Structure Annotation Tool)
MolSurfer (a Macromolecular Interface Navigator)
PIPSA (Protein Interaction Property Similarity Analysis) version 2.0

